



版权说明:本文档由用户提供并上传,收益归属内容提供方,若内容存在侵权,请进行举报或认领
文档简介
电剖析化学导论要点与难点1.电池电动势的正确图示及有关运算2.电极电位的正确表达及有关运算3.有关物理化学常数与电极电位、电池电动势的关系概括电剖析化学是仪器剖析的一个重要分支,是成立在溶液电化学性质基础上的一类剖析方法,或许说利用物质在其溶液中的电化学性质及其变化规律进行剖析的一类方法。电化学性质是指溶液的电学性质(如电导、电量、电流、电位等)与化学性质(如溶液的构成、浓度、形态及某些化学变化等)之间的关系。电剖析化学的分类与特色电剖析化学的分类习惯上按电化学性质参数之间的关系来区分,可分为:电导剖析法、电位剖析法、电解与库仑剖析法、极谱与伏安剖析法等。而往常是区分为三个种类:--以待测物质的浓度在某一特定实验条件下与某些电化学参数间的直接关系为基础的剖析方法。如电导法、电位法、库仑法、极谱与伏安法等。--以滴定过程中,某些电化学参数的突变作为滴定剖析中指示终点的方法(注意:不是用指示剂),如电位滴定、电导滴定、电流滴定等。--经电子作为"积淀剂",使试液中某待测物质经过电极反响转变为固相堆积在电极上、由电极上堆积产物的量进行剖析的方法,如电解剖析法(也称电重量法)。依照IUPAC(国际纯粹和应用化学联合会)1975年的介绍建议,分红三类:--既不波及到双电层,也不波及到电极反响的方法,如电导剖析和高频滴定。--只波及到双电层,但不波及到电极反响的方法,如表面张力法和非法拉第阻抗丈量法。--波及到电极反响的方法,如电位剖析法、电解剖析法、库仑剖析法、极谱和伏安剖析法。本课程学习的是第三类方法电剖析化学的特色--所使用的仪器较简单、小型、价钱较廉价。因丈量的参数为电信号,传达方便,易实现自动化和连续化;--测定快速、简易;--某些新方法的敏捷度高,可作痕量或超痕量剖析,选择性也较好;--不单能够作组分含量剖析,也能够进行价态、形态剖析,还能够作为其余领域科学研究的工具。化学电池化学电池的构成化学电池是化学能与电能相互转变的装置
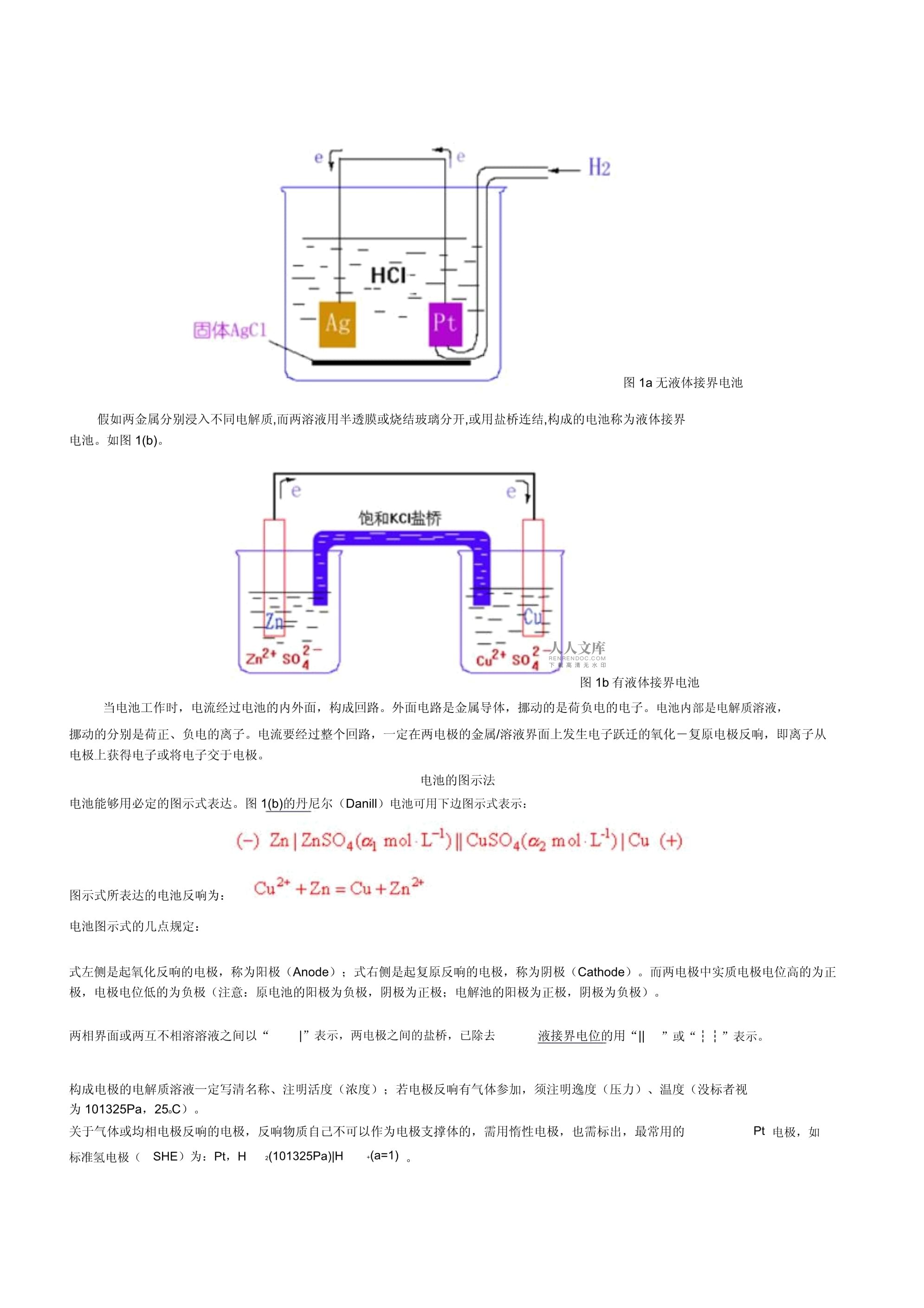
:简单的化学电池由两组金属――电解质溶液系统构成。这种金属――溶液系统称为电极(有时也称半电池),两电极的金属部分经过导线与外电路联络,两电极的溶液部分一定相互交流,以构成一个回路。假如两支构成电极的金属浸入同一个电解质溶液,构成的电池称为无液体接界电极。如图1(a)。图1a无液体接界电池假如两金属分别浸入不同电解质,而两溶液用半透膜或烧结玻璃分开,或用盐桥连结,构成的电池称为液体接界电池。如图1(b)。图1b有液体接界电池当电池工作时,电流经过电池的内外面,构成回路。外面电路是金属导体,挪动的是荷负电的电子。电池内部是电解质溶液,挪动的分别是荷正、负电的离子。电流要经过整个回路,一定在两电极的金属/溶液界面上发生电子跃迁的氧化-复原电极反响,即离子从电极上获得电子或将电子交于电极。电池的图示法电池能够用必定的图示式表达。图1(b)的丹尼尔(Danill)电池可用下边图示式表示:图示式所表达的电池反响为:电池图示式的几点规定:式左侧是起氧化反响的电极,称为阳极(Anode);式右侧是起复原反响的电极,称为阴极(Cathode)。而两电极中实质电极电位高的为正极,电极电位低的为负极(注意:原电池的阳极为负极,阴极为正极;电解池的阳极为正极,阴极为负极)。两相界面或两互不相溶溶液之间以“
|”表示,两电极之间的盐桥,已除去
液接界电位的用“||
”或“┆┆”表示。构成电极的电解质溶液一定写清名称、注明活度(浓度);若电极反响有气体参加,须注明逸度(压力)、温度(没标者视为101325Pa,25oC)。关于气体或均相电极反响的电极,反响物质自己不可以作为电极支撑体的,需用惰性电极,也需标出,最常用的
Pt
电极,如标准氢电极(
SHE)为:Pt,H
2(101325Pa)|H
+(a=1)
。电池的电动势电池的电动势(用Ecell表示,往常简写为E)表示电池的两电极之间的电势差。它包含阴极及阳极的电极电位(分别用或及或表示)及两个半电池电解质溶液的接触电位(称液接界电位,用或表示)液接界电位
的产生--如图
2所示,当两种静态的不同溶液直接接触时,
假如离子不同或离子相同而浓度不同,
由于离子的迁徙(扩散)速度不同,(图中
H+的迁徙速度比
Cl-大)。在其接触界面上产生正负电荷的分别,
因此产生界面的电势差,称为液接界电位,往常简称液接电位。因为难以确立,它影响了电池电动势的丈量,所以一定予除去或尽量降低到最小限度,往常的方法是在两电解质溶液之间用“盐桥”连结,如图3所示。(附:“盐桥”的制作:在饱和的KCl溶液中加入约3%的琼脂,加热使之溶解,趁热倾入U形管中,冷却成凝胶)。“盐桥”的两头插入两电极的溶液中。当“盐桥”与浓度不大的电解质溶液接触时,因为“盐桥”中KCl+-+-低(一般为1-2mv)。图2
液接电位的形成
图
3
液接电位的除去除去
后的电池电动势为:
因为电池图示式中左侧表示阳极,右侧表示阴极。所以电池电动势电池反响自由能变化△G
与电池电动势
E的关系为:(n为反响的电子转移数,F为法拉第常数)在恒温、恒压下,假如自由能降低,△G<0,即E>0,则反响能自觉进行,此电池为原电池;假如自由能高升,△G>0即E<0,则反响不可以自觉进行,若欲使反响进行,需赐予能量,此电池为电解池。电极电位及电极的种类电极电位的形成和表示式1.电极电位的形成以金属与其盐溶液构成的电极为例说明电极电位的形成。金属可当作是由金属离子和自由电子所构成,金属离子以点阵结构有序摆列,电子在此中可自由运动。当金属盐溶液中时,就发生两个相反的过程:一是金属失掉电子生成金属离子而进入溶液,电子留在金属固相中,使金属固相荷负电,而溶液有过多的金属离子而荷正电,这叫金属的溶解压;二是溶液中的金属离子从金属晶格中获取电子生成金属堆积到金属固相上,固相中留有电子空穴而荷正电,而溶液中有剩余的阴离子而荷负电,这叫金属离子的浸透压。两个相反过程的初始速率不会
M插入其均等,这取决于金属及其盐溶液的性质。而不论初始时溶解压大仍是浸透压大,最后溶解压和浸透压都要达到均衡,都在金属与溶液的界面上形成双电层,产生电势差,即产生电极电位。电极电位是一种势垒,其大小是一种相对值,往常以标准氢电极(SHE)作为参照标准:确立某电极的电极电位时,将该电极与SHE构成一个原电池:SHE||待测电极测得该电池的电动势即为该电极的电极电位。(注意:在电位表中,一般以SHE为标准,而在电剖析化学的适用中,常常以饱和甘汞电极SCE为标准)电极电位的表示式电极反响称为半电池反响,半电池反响往常写成复原反响的形式,即:电极电位表示式为:--称为能斯特(Nernst)方程式,在一些书中写为:式中:,分别为半反响中氧化态和复原态物质的活度,称为该电极的标准电极电位。R为气体常数(8.314J/mol
n为单元半反响的电子转移数,·K),F为法拉弟常数(96487C/mol
为,均为),K为热力学温度。
1时的电极电位,当K为278(25oC)时,方程式简化为:应特别指明的是方程式所表示的是电位φ与活度的关系,当有关反响物质的活度均为1时,其电位才是标准电极电,而活度与溶液的离子强度――表现为活度系数有关,而系统均衡时,又会遇到溶液系统的酸效应、络合效应、积淀反响等条件要素的影响,所以标准电极电位在实质应用上有它的限制性。假如考虑上述条件的影响,采纳式量电位(或称条件电位)将更具实质意义。所谓式量电位,即当氧化态和复原态的剖析浓度都是1mol·L-1时的实质电位,以只考虑离子强度的影响为例说明之:=γCγ为活度系数(取决于离子强度)C为摩尔浓度所以:CO=CR=1mol·L-1时,此时有关常数,而
NernstNernst
方程式为:假如其余条件要素存在时,式的对数项中,还应包含其余方程式中的C为剖析浓度(请参阅剖析化学有关内容)。电极的分类可分为两大种类:鉴于电子互换反响的电极和鉴于离子互换或扩散的电极。一般分为五种电极:第一类电极金属与其离子溶液构成系统的电极(活性金属电极),如:银、汞、铜、铅、锌、镉等电极图示式:半反响:电极电位:n+的活度有关.可见φ仅与M第二类电极金属与其难溶盐(或络离子)及难溶盐的阴离子(或配位离子)构成系统的电极,如:银-氯化银电极(Ag/AgCl,Cl-)电极反响:电极电位(25OC):而Ag/Ag+电极的电位为:所以:近似且常用的电极还有甘汞电极-);金属与其络离子构成的电极如银-银氰络离子电极--)。(Hg/Hg2Cl2,Cl(Ag/Ag(CN)2,CN第二类电极的电极电位取决于阴离子的活度,所以能够作为测定阴离子的指示电极;银-氯化银电极及甘汞电极(特别是饱和甘汞电极)又常作为电化学中的二级标准电极。第三类电极金属与两种拥有相同阴离子难溶盐(或难离解络合物)以及第二种难溶盐(或络合物)的阳离子所构成系统的电极。这两种难溶盐(或络合物)中,阴离子相同,而阳离子一种是构成电极的金属的离子,另一种是待测离子。如:等,关于前者电极反响-2+2Ag+CaCO22424因为:所以:关于后者,电极反响:HgY+2e-+Cd2+Hg+CdY能够获取:这种电极能够用于电位滴定中pM的指示电极,在滴定邻近终点时,可视[HgY]/[CdY]基本不变,所以:零类电极惰性金属与可溶性氧化态和复原态溶液(或与气体)构成系统的电极。惰性电极自己不发生电极反响,只起电子转移的介质作用,最常用的是Pt电极。||如:Pt|Fe3+(a1),Fe2+(a2),Pt|Ce4+(a1),Ce3+(a2),氢电极等。膜电极拥有敏感膜并能产生膜电位的电极(鉴于离子互换或扩散的电极)敏感膜指的是对某一种离子拥有敏感响应的膜,其产生的膜电位与响应离子活度之间的关系听从Nernst方程式,整个膜电极的电极电位也听从Nernst方程式。别的,较有适企图义的电极还有:微电极或超微电极Pt丝或玻碳纤维做成的电极,其有好多优秀的特征,用于某些特别的微系统,如生命科学的研究等化学修饰电极(CME)Pt或玻碳电极表面经过共价键合或强吸附或高聚物涂层等方法,把拥有某种功能的基团修饰在电极表面,做成拥有特别性能的电极。参比电极、指示电极、工作电极、协助电极指示电极用来指示电极表面待测离子的活度,在丈量过程中溶液本体浓度不发生变化的系统的电极。如电位丈量的电极,丈量回路中电流几乎为零,电极反响基本上不进行,本体浓度几乎不变。工作电极用来发生所需要的电化学反响或响应激发信号,在丈量过程中溶液本体浓度发生变化的系统的电极。如电解剖析中的阴极等。参比电极用来供给标准电位,电位不随丈量系统的组分及浓度变化而变化的电极。这种电极一定有较好的可逆性、重现性和稳固性。常用的参比电极有SHE、Ag/AgCl、Hg/Hg2Cl2电极,尤以甘汞电极(SCE)使用得最多。Hg/Hg2Cl2电极的φ取决于aCl-,25oC时CKCl(mol·L-1)0.11.0饱和φ(V)+0.3365+0.2828+0.2438协助电极--或称对电极在电化学剖析或研究工作中,常常使用三电极系统,除了工作电极,参比电极外,还需第三支电极,此电极所发生的电化学反响并不是测示或研究所需要的,电极仅作为电子传达的场所以便和工作电极构成电流回路,这种电极称为协助电极或对电极。去极化与极化电极去极化电极在电化学丈量中,电极电位不随外加电压的变化而变化,或当电极电位改变很小时所产生的电流改变很大的电极。如饱和甘汞电极、电位剖析法中的离子选择电极均为去极化电极。极化电极在电化学丈量中,电极电位随外加电压的变化而变化,或当电极电位改变很大时所产生的电流改变很小的电极。极化电极被极化时,电极电位将偏离均衡系统的电位,偏离值称为过电位。如电解、厍仑剖析中的工作电极及极谱剖析法中的指示电极都是极化电极。产生极化的原由(主要有两种)浓差极化可逆且快速的电极反响使电极表面液层内反响离子的浓度快速降低(或高升)--->电极表面与溶液本体之间的反响离子浓度不相同,形成必定的浓度梯度--->产生浓差极化--->电极表面液层的离子浓度决定了电极的电位,此电位偏离了电极的均衡电位,偏离值称为浓差过电位。电化学极化电极的反响速度较慢―――>当电流密度较大时,惹起电极上电荷的积累―――>产生电化学极化―――>电极的电位取决于电极上所积累的电荷,此电位偏离了电极的均衡电位,偏离值称为活化过电位。不论是哪一种极化,都使阴极的电位较均衡电位为负,使阳极的电位较均衡电位为正。阴极过电位ηc=φc-φrc(负值)阳极过电位ηa=φa-φra(正当)φ为实质电位,φr为均衡电位电池总过电位η=φa-φc(两电极过电位绝对电位值之和)思虑题与练习题化学电池由哪几部分构成?如何表达电池的图示式?电池的图示式有哪些规定?电池的阳极和阴极,正极和负极是如何定义的?阳极就是正极,阴极就是负极的说法对吗?为何?电池中"盐桥"的作用是什么?盐桥中的电解质溶液应有什么要求?电极电位及电池电动势的表示式如何表示?应注意什么问题?何谓式量电位?如何表示?电极有几种种类?各样种类电极的电极电位如何表示?何谓指示电极、工作电极、参比电极和协助电极?何谓电极的极化?产生电极极化的原由有哪些?极化过电位如何表示?8.写出以下电池的半电池反响和电池反响,计算电动势。这些电池是原电池仍是电解池?极性如何?(设T为25℃,活度系数均为1)(1)Pt│Cr3+(1.0×10-4mol·L-1),Cr2+(0.10mol·L-1)‖Pb2+(8.0×10-2mol·L-1)│Pb已知:答案:E=0.429V,是原电池(2)已知:答案:E=0.007V,是原电池(3)Pt,H2(20265Pa)│HCl(0.100mol·L-1-1)‖HClO4(0.100mol·L)│Cl2(50663Pa),Pt已知:答案:E=1.448V,是原电池(4)Bi│BiO+(8.0×10-2mol·L-1),H+(1.00×10-2mol·L-1)‖I-(0.100mol·L-1),AgI(饱和)│Ag已知:答案:E=-0.261V,是电解池已知以下半电池反响及其标准电极电位:Sb+3H++3e-=SbH39.计算以下半电池反响--值答案:-1.34VSb+3H2O+3e=SbH3+3OH在25℃时的10.电池:Hg│Hg2Cl2,Cl-(饱和)‖Mn+│M在25℃时的电动势为0.100V;当Mn+的浓度稀释为本来的1/50时,电池的电动势为0.050V。试求电池右侧半电池反响的电子转移数(n值)。答案:n=211.以下电池的电动势为0.693V(25℃)Pt,H2(101325Pa)│HA(0.200mol·L-1),NaA(0.300mol·L-1)‖SCE,不考虑离子强度的影响,请计算HA的离解常数Ka。答案:Ka=3.9×10以下电池的电动势为-0.138V(298K)Hg│Hg(NO3)2(1.0×10-3mol·L-1),KI(1.00×10-2mol·L-1)‖SCE试计算:的配合稳固常数K。答案:K=6.5×1029已知以下半电池反响及其标准电极电位:试计算:的配合稳固常数K答案:6714.以下电池的电动势为0.411V(25℃)Pb│PbCl2(饱和),Cl-(0.020mol·L-1)‖SCE已知,不考虑离子强度的影响。请计算PbCl2的KSP。答案:Ksp=1.7×10-515.有一电池:Zn│Zn2+(0.0100mol·L-1)‖Ag+(0.300mol·L-1)│Ag计算该电池298K时的电动势为多少?当电池反响达到均衡外线路无电子流经过时,Ag+浓度为多少?已知:答案:E=1.59V,浓度=1.55×10-27mol·L-1电位剖析法要点与难点1.膜电位及离子选择电极电位的产生及表达式2.各种离子选择性电极的响应机理,pH玻璃电极尤其重要3.离子选择性电极的特征参数,尤以电位选择系数为重要4.离子选择性电极的应用电位剖析法的基根源理电位剖析法是利用电极电位与溶液中待测物质离子的活度(或浓度)的关系进行剖析的一种电化学剖析法。Nernst方程式就是表示电极电位与离子的活度(或浓度)的关系式,所以Nernst方程式是电位剖析法的理论基础。电位剖析表示图电位剖析法利用一支指示电极(对待测离子响应的电极)及一支参比电极(常用SCE)构成一个丈量电池(是一个原电池)如上图所示。在溶液均衡系统不发生变化及电池回路零电流条件下,测得电池的电动势(或指示电极的电位)E=φ参比-φ指示
因为φ参比不变,φ指示切合
Nernst
方程式,所以
E的大小取决于待测物质离子的活度(或浓度),从而达到剖析的目的。电位剖析法的分类和特色电位剖析法的分类直接电位法――利用专用的指示电极――离子选择性电极,选择性地把待测离子的活度(或浓度)转变为电极电位加以丈量,依据Nernst方程式,求出待测离子的活度(或浓度),也称为离子选择电极法。这是二十世纪七十年月初才发展起来的一种应用宽泛的快速剖析方法。电位滴定法――利用指示电极在滴定过程中电位的变化及化学计量点邻近电位的突跃来确立滴定终点的滴定剖析方法。电位滴定法与一般的滴定剖析法的根本差异在于确立终点的方法不同。电位剖析法的特色★直接电位法应用范围广――可用于很多阴离子、阳离子、有机物离子的测定,特别是一些其余方法较难测定的碱金属、碱土金属离子、一价阴离子及气体的测定。因为测定的是离子的活度,所以能够用于化学均衡、动力学、电化学理论的研究及热力学常数的测定。测定速度快,测定的离子浓度范围宽。能够制作成传感器,用于工业生产流程或环境监测的自动检测;能够微型化,做成微电极,用于微区、血液、活体、细胞等对象的剖析。★电位滴定法1正确度比指示剂滴定法高,更适合于较稀浓度的溶液的滴定。2可用于指示剂法难进行的滴定,如极弱酸、碱的滴定,络合物稳固常数较小的滴定,浑浊、有色溶液的滴定等。3可较好地应用于非水滴定。离子选择性电极的基本结构离子选择性电极(Ion-SelectiveElectrode,ISE)是其电极电位对离子拥有选择性响应的一类电极,它是一种电化学传感器,敏感膜是其主要构成部分,其基本结构见图1。ISE由四个基本部分构成:电极腔体――玻璃或高分子聚合物资料做成内参比电极――往常为Ag/AgCl电极内参比溶液――由氯化物及响应离子的强电解质溶液构成敏感膜――对离子拥有高选择性的响应膜图1离子选择性电极表示图离子选择性电极的电极电位1.膜电位扩散电位φd――两种不同离子或离子相同而活度不同的溶液,其液液界面上因为离子的扩散速度不同,能形成液接电位,也可称扩散电位。离子的扩散属于自由扩散,没有强迫性和选择性,正、负离子均可进行。扩散电位不单存在于液液界面,也存在于固体内部。在离子选择性电极的敏感膜中也可产生扩散电位。扩散电位与离子的迁徙数ti有关,当扩散阳离子和阴离子的迁徙数(t+、t-)相同时,φd=0。关于一价离子φd可表示为:(分别表示同一离子在两种溶液中的活度)道南(Donnan)电位φD――若有一种带负电荷载体的膜(阳离子互换物质)或选择性浸透膜。它能发生互换或只让被选择的离子经过,当膜与溶液接触时,膜相中可活动的阳离子的活度比溶液中高,或许只同意阳离子经过,而阻挡阴离子经过,最后结果造成液和膜两相界面上正、负电荷散布不均匀,形成双电层结构而产生电势差。这种电荷的迁徙形式带有选择性或强迫性,产生的电位是相间电位,称为道南电位。(、分别为离子在溶液中及膜界面上的活度)注意:式中""号表示阳离子为"+",阴离子为"-"。ISE的膜电位φMφM包含液、膜两相界面离子扩散或互换所产生的道南电位φD及膜中内、外两表面间隔子扩散产生的扩散电位φd,如图2所示:(式中z为离子的电荷数)因为膜的响应拥有选择性,可以为只有一种阳离子或阴离子能进行互换或扩散。关于阳离子来说,t-=0,t+=1,则为定值所以对阴离子来说,图2膜电位的产生3.ISE的电极电位φISE是定值所以:(阳离子为"+",阴离子为"-")可见φISE取决于待测离子的活度,所以上式也称为ISE的Nernst方程,它是ISE剖析法的依照。K也能够写成,它包含、内膜界面电位、膜内扩散电位、膜不对称电位等。离子选择性电极的分类(注:原电极是指敏感膜直接与测定试液接触的ISE,敏化ISE是以原电极为基础装置成的选择电极。)晶体膜电极晶体膜电极分为均相、非均相晶膜电极。均相晶膜由一种化合物的单晶或几种化合物混淆均匀的多晶压片而成。非均相膜由多晶中掺惰性物质经热压制成。晶体膜电极结构如图3所示([b]为全固态型电极)。图3晶体膜电极的结构晶体膜电极的响应机理包含两个方面:★晶膜表面与溶液两相界面上响应离子的扩散形成界面电位(道南电位)――响应离子进入晶体中可能存在的晶格离子空穴,而晶膜中的晶格离子也会扩散进入溶液而在膜中留下空穴,均衡时在界面上形成双电层而产生电位。★晶膜内部离子的导电体制形成了扩散电位――因为膜、液界面上响应离子的扩散,使膜内晶格离子散布不均匀,即空穴不均匀,惹起晶格离子的扩散,-的扩散LaF3+空穴→++-必空穴的挪动,如LaF3晶体中FLaF2(新空穴)F须指出的是:能传达的电荷不过少量晶格能小的晶体,并且只好是半径最小、电荷最少的晶格离子才能扩散挪动。如LaF3中F-。扩散的结果产生了扩散电位。此类电极的扰乱是共存离子与晶格离子生成难溶盐或稳固的络合物,改变晶膜表面的性质,而不是共存离子进入膜参加--响应。如OH对F电极的扰乱是产生La(OH)3积淀所致。所以,晶膜电极的选择性取决于膜化合物和共存离子与晶格离子生成化合物溶解度的相对大小,而检测限取决于膜化合物的Ksp。晶体膜电极常用的有:氟电极——膜为LaF3单晶片,掺入少许Eu2+、Ca2+以改良其导电性能。结构如图4所示:电极电位丈量电池( )Ag|AgCl,Cl-(0.1mol·L-1),F-(0.1mol·L-1)|试液(aF-)||Cl-(饱和),Hg2Cl2|Hg(SCE)氟电极使用的酸度范围为pH5.0~5.5图4氟离子选择电极硫离子选择电极――膜为Ag2S粉末压片制成,膜内Ag+是电荷的传达者测定含S2-溶液时,氯、溴、碘离子选择电极――膜分别由AgCl、AgBr、AgI与Ag2S粉末混匀压片制成。膜内的电荷也是由Ag+传达。铜、铅、镉离子选择电极――膜是分别由Cus、PbS、CdS与Ag2S粉末混匀压片制成。膜内的电荷仍旧由+2+不Ag传达,M参加传达电荷。表1列出常用的晶体膜电极表1晶体电极的品种和性能电极膜资料线形响应浓度范围合用主要扰乱离子C/mol.L-1pH范围F-2+5*10-7~1*10-15~6.5-LaF3+EuOHCl-AgCl+Ag2S5*10-5~1*10-12~12Br-,S2O32-,I-,CN-,S2-Br-AgBr+Ag2S5*10-6~1*10-12~12S2O32-,I-,CN-,S2-I-AgI+Ag2S1*10-7~1*10-12~11S2--AgI1*10-6~1*10-1>10I-CNAg+,S2-Ag2S1*10-7~1*10-12~12Hg2+2+CuS+AgS5*10-7~1*10-12~10+2+,Fe3+,Cl-CuAg,Hg2Pb2+PbS+Ag2S5*10-7~1*10-13~6Cd2+,Ag+,Hg2+,Cu2+,Fe3+,Cl-Cd2+PbS+Ag2S5*10-7~1*10-13~10Pb2+,Ag+,Hg2+,Cu2+,Fe3+玻璃电极1.玻璃电极的结构及种类1.玻璃电极的结构及种类玻璃电极包含对H+响应的pH玻璃电极及对K+、Na+离子响应的pK、pNa玻璃电极。玻璃电极的结构相同由电极腔体(玻璃管)、内参比溶液、内参比电极及敏感玻璃膜构成,而要点部分为敏感玻璃膜。pH玻璃电极的结构如图5所示。此刻不少商品的pH玻璃电极制成复合电极,它集指示电极和外参比电极于一体,使用起来甚为方便和牢靠。玻璃电极依照玻璃球膜资料的特定配方不同,能够做成对不同离子响应的电极。如常用的以考宁015玻璃做成的pH玻璃电极,其配方为:Na2O21.4%,CaO6.4%,SiO272.2%(摩尔百分比),其pH丈量范围为pH1-10,若加入必定比率的Li2O,能够扩大丈量范围。改变玻璃的某些成分,如加入必定量的Al2O3,能够做成某些阳离子电极,如表2所示。图5PH玻璃电极表2阳离子玻璃电极主要响应离子玻璃膜构成(摩尔分数,10-2)选择性系数Na2OAl2O3SiO2Na+111871K+3.3×10-3(pH7),3.6×10-4(pH11),Ag+500K+27568Na+5×10-2Ag+111871Na+1×10-328.819.152.1H+1×10-5Na+0.3Li+Li2O152560K+<1×10-3pH玻璃电极的响应机理硅酸盐玻璃的结构――玻璃中含有金属离子、氧和硅,Si-O键在空间中构成固定的带负电荷的三维网络骨架,金属离子与氧原子以离子键的形式联合,存在并活动于网络之中担当着电荷的传导,其结构如图6所示。图6硅酸盐玻璃的结构感敏玻璃膜水化胶层的形成――新做成的电极,+所占有。当玻璃膜与纯水或稀酸接触时,因为干玻璃膜的网络中由Na++的结协力,因此发生了以下的互换反响-++-++Si-O与H的结协力远大于与NaGNa+HGH+Na反响的平衡常数很大,向右反响的趋向大,玻璃膜表面形成了水化胶层。所以水中浸泡后的玻璃膜由三部分构成:膜内外两表面的两个水化胶层及膜中间的干玻璃层,如图7所示。pH玻璃电极的膜电位及电极电位――形成水化胶层后的电极浸入待测试液中时,在玻璃膜内外界面与溶液之间均产生界面电位,而在内、外水化胶层中均产生扩散电位,膜电位是这四部分电位的总和。即:图7玻璃膜的水化胶层及膜电位的产生Baucke以为水化胶层中的离解均衡及水化胶层中++的互换是决定界面电位的主要要素,即:H与溶液中H并且,当玻璃膜内表面面的性状相同,能够认为:所以:则pH玻璃电极的电极电位为:pH玻璃电极的不对称电位φ不――依照上边推得的膜电位公式,当膜内外的溶液相同时,φM=0,但实质上仍有一很小的电位存在,称为不对称电位,其产生的原由是因为膜的内表面面的性状不行能完好相同,即与、φd内与φd外不同惹起的。影响它的要素主要有:制作电极时玻璃膜内表面面产生的表面张力不同,使用时膜内表面面所受的机械磨损及化学吸附、浸蚀不同。不同电极或同一电极使用状况、使用时间不同,都会使φ不不相同,所以φ不难以丈量和确立。干的玻璃电极使用前经长时间在纯水或稀酸中浸泡,以形成稳固的水化胶层,可降低
φ不
;pH丈量时,先用
pH标准缓冲溶液对仪器进行定位,可除去
φ不
对测定的影响。各样离子选择电极均存在不同程度的
φ不
,而玻璃电极较为突出。pH玻璃电极的
"钠差"和"酸差""钠差"――当丈量
pH较高或
Na+浓度较大的溶液时,测得的
pH值偏低,称为
"钠差"或"碱差"。每一支
pH玻璃电极都有一个测定
pH高限,高出此高限时,
"钠差"就展现了。产生
"钠差"的原由是
Na+参加响应。"酸差"――当丈量
pH小于
1的强酸、或盐度大、或某些非水溶液时,测得的
pH值偏高,称为
"酸差"。产生"酸差"的原因是:当测定酸度大的溶液时,玻璃膜表面可能吸附H+,当测定盐度大或非水溶液时,溶液中变小。3.pH的适用(操作性)定义及pH的丈量+pH的热力学定义为:活度系数γH难以正确测定,此定义难以与实验测定值严格有关。所以提出了一个与实验测定值严格有关的适用(操作性)定义。以下的丈量电池:所以缓冲溶液(pH
K'是一个不确立的常数,所以不可以经过测定E直接求算S)及试液(pHX)的电动势(ES及EX),获取:
pH,而是经过与标准
pH
缓冲溶液进行比测,分别测定标准即pH值是试液和pH标准缓冲溶液之间电动势差的函数,这就是pH的适用(操作性)定义。美国国家标准局已确立了七种pH标准溶液。我们常用的三种标准溶液为:邻苯二甲酸氢钾、磷酸二氢钾-磷酸一氢钾、硼砂,25℃时的pH分别为4.01、6.86、9.18。实质工作中,用pH计丈量pH值时,先用pH标准溶液对仪器进行定位,而后丈量试液,从仪表上直接读出试液的pH值。流动载体电极流动载体电极--也称液膜电极,其结构如图8所示。液体敏感膜由三部份构成:电活性物质(载体)、有机溶剂(可溶解载体,也是增塑剂)、支撑膜(常用PVC塑料、垂玻璃、聚四氟乙烯微孔膜)。此中最重要的是电活性载体,依据其性质有三种种类:图8液膜电位阳性――带正电荷载体,若有机大阳离子、鎓类(季铵、季磷、季砷类)离子、络阳离子、碱性染料等,这种载体能响应无机、有机阴离子或络阴离子,如NO3-选择电极等。阴性――带负电荷载体,若有机大阴离子、羧基等,可响应阳离子,如2+Ca选择电极,一些药物电极等。中性――载体为一些拥有未成键电子(n电子)的中性大分子螯合剂,如某些抗生素、冠醚化合物及开链酰胺等,可响应阳离子,如K+选择电极。当液膜电极与丈量溶液接触时,响应离子能够在液、膜两相中自由出入(互换、扩散),进入膜相中的响应离子与约束在膜相中的电活性物质联合成离子型的缔合物或络合物,相同被约束在膜相中,而响应离子的陪伴离子不能进入膜内,以以下图所示,因为响应离子在液、膜两相间的互换及在膜相中的扩散,就形成了膜电位。表3、表4列出几种常有流动载体电极。液膜电极膜电位产生表示图表3带电荷的流动载体电极线形响应浓度范围离子电极活性物质主要扰乱离子C/mol.L-1Ca2+水硬度2+2+(Ca+Mg)NO3-ClO4-BF4-离子电极K+
二(正辛基苯基)磷酸钙溶于1×10-5~1×10-1Zn2+,Mn2+,Cu2+苯基磷酸二辛酯二癸基磷酸钙溶于癸醇1×10-5~1×10-1Na+,K+,Ba2+,Sr2+Cu2+,Ni2+,Zn2+,Fe2+四(十二烷基)硝酸铵5×10-6~1×10-1--,I-,ClO-42邻二氮杂菲铁(Ⅱ)配合物1×10-5~1×10-1-OH三庚基十二烷基氟硼酸铵1×10-6~1×10-1I-,SCN-,ClO4-表4中性载体电极中性载体线形响应浓度范围主要扰乱离子C/mol.L-1颉氨霉素1×10-5~1×10-1Rb+,Cs+,NH4+二甲基二苯基,30-冠醚-101×10-5~1×10-1Rb+,Cs+,NH4+Na+三甘酰双苄苯胺1×10-4~1×10-1K+,Li+,NH4+四甲氧苯基,24-冠醚-81×10-5~1×10-1K+,Cs+Li+开链酰胺1×10-5~1×10-1K+,Cs++类放线菌素+甲基类放线菌素1×10-5~1×10-1++NH4K,RbBa2+四甘酰双二苯胺5×10-6~1×10-1K+,Sr2+气敏电极气敏电极是一种气体传感器,由离子选择电极(如pH电极等)作为指示电极,与外参比电极一同插入电极管中构成复合电极,电极管中充有特定的电解质溶液――称为中介液,电极管端部紧靠离子选择电极敏感膜处用特别的透气膜或缝隙间隔把中介液与外测定液分开,构成了气敏电极。结构如图9,10所示。图9隔阂式气敏电极图10气隙式气敏电极丈量时,试样中的气体经过透气膜或缝隙进入中介液并发生作用,惹起中介液中某化学均衡的挪动,使得能惹起选择电极响应的离子的活度发生变化,电极电位也发生变化,进而能够指示试样中气体的分压。如氨气敏电极,以pH玻璃电极为指示电极,透气膜为聚偏四氟乙烯,中介质为NH4Cl溶液,NH3穿过透气膜进入NH4Cl溶液,惹起以下均衡的挪动:表5气敏电极的品种及性能气敏离子指检测限试液中介溶液化学反响均衡-1pH扰乱电极示电极C/mol.LCO2H+0.01mol.L-1NaHCO310-5<4+10-6>11挥发性34NHHNHCl胺NO+NaNO5×10-7柠檬酸缓冲SO,CO222液2SO+NaHSO10-6-Cl,NO22342H2SS2-柠檬酸缓冲液pH510-8<5O2HCNAg+KAg(CN)210-7<7H2SHFF-H+10-3<2HAcH+NaAc10-3<2Cl2Cl-5×10-3<2HSO4缓冲液生物电极酶电极――在指示电极(离子选择性电极或电流型传感电极)上覆盖一层活性酶物质,经过酶的酶促作用,使待测物质(底物)反响生成指示电极能响应的物质,进而达到间接测定的目的。如尿素的测定:表6列出几种常有的酶电极。组织电极――利用动、植物组织内存在的某种酶,将这些物质覆盖在指示电极上,能够做成组织电极。如将猪肝切片夹在尼龙网中紧贴在氨气敏电极上,猪肝组织中的谷氨酰胺酶能催化谷氨酰胺反响开释出氨,进而能够测定试样中的谷氨酰胺。又如将香蕉与碳糊混淆贴在氧电极上,能够测定多巴胺的含量。表7列出几种组织电极。表6酶电极的构成和性能测定物质酶指示电极测定范围或检测物mol.L-1葡萄糖葡萄糖氧化酶O21×10-4~2×10-2脲脲酶NH31×10-5~1×10-2胆固醇胆固醇氧化酶H2O21×10-5~1×10-2L-谷氨酸谷氨酸脱氢酶+1×10-4~1×10-24L-赖氨酸赖氨酸脱羧酶CO21×10-4~1×10-2表7组织电极组织测定对象使用指示电极猪肾谷氨酰胺NH3兔肝乌嘌呤NH3黄瓜谷氨酸CO2大豆尿素NH、CO32香蕉肉多巴胺O2离子敏感场效应晶体管(ISFET)图11场效应晶体管的结构ISFET是一种微电子化学敏感元件,它既有ISE对敏感离子响应的特征,又保存场效应晶体管的性能,是ISE制造工艺和半导体微电子技术联合的产物。图11表示一般金属-氧化物-半导体场效应晶体管(MOSFET)的结构及工作原理,漏流Id的大小受栅极g与源极s之间的电压Ugs控制,并为栅压和源极与漏极d间电压Uds的函数。用ISE的敏感膜取代MOSFET的金属栅极时,即成为ISFET,当它与试液接触并与参比电极构成丈量系统时,电池的电动势加到源-栅极之间,即膜电位叠加在栅压上,产生的Id将与响应离子活度之间有近似于Nernst方程式的关系。ISFET拥有好多长处,如能够做成全固态器件,体积小,易微型化和多功能化,自己拥有高阻抗变换和放大功能。但制造工艺复杂,绝缘要求高。离子选择微电极将往常的ISE的敏感膜微型化即成为离子选择微电极,当前已制成有H+、Na+、K+和Cl-等微电极。把玻璃管尾端拉制成直径为2~20μm的毛细管,经疏水办理此后,将毛细管在溶有电活性物质的溶液中蘸取一下,等溶剂挥发后,玻璃管中再配上内参比电极、内参比溶液即成。可见它的结构与液膜电极近似。这种电极对展开生化、生理及医学临床研究是很实用的。离子选择性电极的特征参数1.Nernst响应、线性范围和检测下限-----电极校准曲线φISE随离子活度变化的特征称为响应,若这种响应切合Nernst方程式,这称为Nernst响应:在实质丈量中,以φ对lgα(或pα)作图,所得的曲线称为校订曲线,对阳离子来说,如上图所示。可见当待测离子的活度降低到某必定值时,曲线开始偏离Nernst方程的线性。校订曲线的直线部分所对应的离子活度范围称为ISE响应的线性范围。直线部分与水平部分延长线的交点所对应的离子活度称为ISE的检测下限。2.响应斜率校订曲线线性响应部分的直线斜率,称为ISE的实质响应斜率,S也称为级差。依照Nerst响应,直线斜率应为,时为,称为ISE的理论响应斜率S理。S一般比S理小,,称为变换系数,≤1,其越靠近1越好。当时,就一般以为ISE的敏捷度过低而不宜使用。3.电位选择系数任何一个ISE对一特定离子的响应都不会是绝对专一的,溶液中的某些共存离子可能也会有响应,即共存离子对ISE的电极电位也有贡献,此时的电位方程式应表达为:式中i为主响应离子,j,k为共存离子,此式称为Nernst扩大式(或称修正式)、称为ISE的电位选择系数。它的大小表示ISE抵挡其余扰乱离子的能力,能够说,是当待测离子i与扰乱离子j所贡献的电位相同时,i离子的活度与j离子的活度的比值(离子电荷相同时),所以越小,ISE的选择性越好。如关于Zi=Zj,当=10-2时,表示ISE对i的响应比对j的响应敏捷100倍。借助选择系数,能够预计扰乱离子对待测离子的丈量结果所产生的偏差:偏差%=例:NO3-ISE的,在0.50mol/L的Na2SO4中测定8.2×10-4mol/L的NO3-,由SO42-惹起的测定偏差为:可从有关手册中查的,但严格地说,它并不是完好部是一个常数,与丈量的条件有关。测定电位选择系数的方法有:分别溶液法--分别配制活度相同的主响应离子i和扰乱离子j的标准溶液,分别测其电位:假如阳离子得式中S为ISE的实质响应斜率两式相减得关于同价离子关于异价离子图12
固定扰乱法测定选择系数混淆溶液法--它是在主响应离子
i和扰乱离子j共存时求测选择系数,有固定扰乱法和固定主响应离子法。固定扰乱法为:配制一系列固定
αj而改变αi的混淆溶液,分别测定电位φi作φi~lgαi曲线,如图
12阳离子
ABCD线,CD直线段表示αi>>αj,j
离子的影响可忽视,则AB直线段表示,αi
<<αj
,电位几乎由αj决定,i
离子的响应能够忽视,则
AB、CD延长线交于
M,所对应的
i活度为
,
=φj所以4.响应时间ISE的实质响应时间是指从ISE和参比电极一同接触丈量溶液到电极电位数值稳固(颠簸在1mV之内)所经历的时间。影响响应时间的要素有:ISE膜电位均衡的时间(膜性能)、参比电极的稳固性、搅拌速度以及响应离子的性质、介质条件、温度等。丈量时,常用搅拌丈量溶液来缩短ISE的响应时间。5.温度系数和等电位点温度系数是电位关于温度的变化率,即(由三项构成)
。所以第一项为哪一项电极系统的标准电位系数,温度对膜资料、内参比系统、不对称电位的影响。第二项是Nernst方程式斜率温度系数,热力学上所固有的。第三项是溶液特征温度系数,温度对体积、活度等的影响,此项较小,一般可忽视。关于每个ISE来说,有一个电位不随温度变化而变化的点,即=0,叫等电位点。若略去第三项,则等电位点能够经过改变内参比系统来实现,经过设计,使之在待测离子活度值的邻近,可使温度对电位的影响降低。其余特征内阻――包含膜内阻及内参比系统内阻,膜内阻是主要的,晶体膜内阻较低,为兆欧级;流动载体膜内阻高,为十兆欧级;玻璃膜内阻最高,达百兆欧级。因为电极内阻高,所以配用的仪器(离子计)需要高输入阻抗,一般1011Ω以上。稳固性――电位漂移状况,与膜稳固性、电极结构、绝缘性能有关。别的还有重现性及寿命等特征。丈量仪器与参比电极电位剖析法的丈量电池由离子选择电极、参比电极及待测溶液构成,用磁力搅拌器搅拌丈量溶液,用高输入阻抗的丈量仪器丈量电动势,如图13所示图13丈量电池特别指出的是:丈量电动势的仪器一定是高输入阻抗的电子伏特计。这是因为ISE的内阻极高,尤以玻璃电极最高,达108Ω。若不是采纳高输入阻抗的丈量仪器,当有极细小的电流(如10-9A)经过回路时,在内阻108Ω的电极上电位降达0.1V,造成pH丈量偏差近2pH单位。可用图14表示电极内阻R电极及丈量仪器输入阻抗R仪器的关系,而用下式预计丈量仪器所需的输入阻抗丈量偏差%=比方要达到0.1%的丈量偏差,关于内阻108Ω的电极来说,仪器输入阻抗需1011Ω。参比电极:要求:可逆性、重现性、稳固性;种类:氢电极、Ag/AgCl、Hg/Hg2Cl2(甘汞电极);最常使用的是饱和甘汞电极(SCE)。该电极25℃时电位为0.2438V,稳固性很好,但使用温度不可以超出80℃。否Hg2Cl2发生歧化反响。甘汞电极丈量时温度滞后现象较为严重。图14丈量仪器的输入阻抗活度与浓度Nernst方程式表示的是电极电位与离子活度之间的关系式,而关于剖析化学来说,测定的是离子浓度而不是活度,活度与浓度的关系为:γ为活度系数,由溶液的离子强度所决定。所以电位方程式变换为:为此,在系列的丈量中一定使γ基本不变,才不会影响测定系列的结果,在电位剖析法中经过加入总离子强度调理缓冲剂(TISAB)来实现的。在实质工作中,TISAB所含的成分有三个作用:控制必定的离子强度,控制必定的丈量酸度,含有络合剂可遮蔽扰乱离子。直接电位法1.直接比较法--也称直读法如丈量离子A,构成电池为:则测定标准溶液
pAs的电动势
Es,再测未知溶液
pAx的电动势
Ex获取
先(阳离子为+,阴离子为-)在实质丈量中,需用两个不同浓度的标准溶液
pAs1、pAs2,且
pAs1<pAx<pAs2,分别用两个标准溶液对离子计进行斜率校订及定位,而后测定未知溶液,从离子计上直接读出
pAx值。标准曲线法--适于大量量且构成较为简单的试样剖析配制一系列(一般为5个)与试样溶液构成相像的标准溶液Ci,与试样溶液相同加入TISAB,分别丈量E(或φ)。绘制E~lgCi(或E~pCi)标准曲线,由未知试样溶液所测的Ex从曲线中求得Cx。3.标准加入法--也称增添法将小体积――Vs(一般为试液的1/50~1/100)而大浓度――Cs(一般为试液的100~50倍)的待测组分标准溶液,加入到必定体积的试样溶液中,分别丈量标准加入前后的电动势,进而求出Cx。可分为单次标准加入法和连续标准加入法两种。(1)单次标准加入法依照上述丈量电池的构成图示式,关于阳离子的丈量来说先丈量体积为Vx的试样溶液的电动势Ex,则,再加入浓度为Cs,体积为Vs的标准溶液后测定,则(标准加入前后丈量溶液的组份基本不变)则整理后获取(S为ISE的实质响应斜率,而非)当Vs<<Vx时,假如测定阴离子,则,而Cx的结果表达式和上式相同。所以,测定阴阳离子的统一式为连续标准加入法--格兰(Gran)作图法在测定过程中,连续多次(3~5次)加入标准溶液,多次测定E值,依照上述电池的图示式,关于阴离子丈量来说,每次E值为变换整理后得:所以与成线性关系。图15连续标准加入法曲线每次加入Vs(累加值),测出一个E值,并计算出的值,绘制~Vs曲线,如图2.15所示,延长直线交于Vs轴的Vs'(呈负值),即:也就是:所以:(关于阳离子,则前面函数式中指数项的指数为负值,即及,其余不变)上述的计算是很复杂的,1969年格兰作图法用于直接电位法。格兰作图法是采纳一种半反对数的格兰坐标纸,直接作E~Vs曲线,结果的计算公式同上。使用格兰坐标纸时要注意到:――坐标纸的纵坐标是以10%体积变化率校订的半反对数,规定一价离子的响应斜率为58mV,而10E/S以105/58为单位设计,即纵坐标每大格为5mV。――横坐标以Vx取100mV设计,每大格为1mL(若Vx取50mL,每格Vs为0.5mL)。――若ISE的响应斜率S不是58mV,应进行空白试验。格兰作图法如图16所示直接电位法的偏差16Gran作图法直接电位法测定浓度结果的偏差主要由电动势E的丈量偏差惹起的。或则相对偏差或(E的单位为伏特)若E丈量偏差为±0.1mv时,测定一价离子的浓度相对偏差为±0.4%,二价离子为±0.8%。电位滴定法的原理和装置图17电位滴定基本仪器装置电位滴定法与直接电位法的不同在于,它是以丈量滴定过程中指示电极的电极电位(或电池电动势)的变化为基础的一类滴定剖析方法。滴定过程中,跟着滴定剂的加入,发生化学反响,待测离子或与之有关的离子活度(浓度)发生变化,指示电极的电极电位(或电池电动势)也跟着发生变化,在化学计量点邻近,电位(或电动势)发生突跃,由此确立滴定的终点。所以电位滴定法与一般滴定剖析法的根本不同是确立终点的方法不同。电位滴定法的装置由四部分构成,即电池、搅拌器、丈量仪表、滴定装置,如图
17所示。滴定终点确实定滴定终点确实定:有作图法和二级微商计算法两种1.作图法――作φ~V曲线(即一般的滴定曲线),以测得的电位φ(或电动势E)对滴定的体积V作图获取图18(a)的曲线,曲线的突跃点(拐点)所对应的体积为终点的滴定体积Ve。――作△φ/△V~V曲线(即一级微分曲线),关于滴定突跃较小或计量点前后滴定曲线不对称的,能够用△φ/△V(或△E/△V)对△V相应的两体积的均匀值(即作图,获取图18(b)的曲线,曲线极大值所对应的体积为
)Ve。――作△22~V曲线(即二级微商曲线),以△22φ/△Vφ/△V(或△2E/△V2)对二次体积的均匀值(即)作图,获取图18(c)曲线,曲线与V轴交点,即△22=0φ/△V所对应的体积为Ve。――作△V/△φ~V曲线,只需在计量点前后取几对数据,以△V/△φ对V作图,可获取两条直线,图18(d)所示,其交点所对应的体积为Ve。2.二级微商计算法从二级微商曲线可见,当△22的两个相邻值出现相反符号时,两个滴定体积V1,V2之间,必有△22的一φ/△Vφ/△V=0点,该点对应的体积为Ve。用线性内插法求得φe、Ve:自动电位的滴定从前及当前还有许多使用自动电位滴定的装置如图19所示。在滴定管尾端连结可经过电磁阀的细乳胶管,此管下端接上毛细管。滴定前依据详细的滴定对象为仪器设置电位(或pH)的终点控制值(理论计算值或滴定实验值)。滴定开始时,电位丈量信号使电磁阀断续开关,滴定自动进行。电位丈量值抵达仪器设定值时,电磁阀自动封闭,滴定停止。现代的自动电位滴定已宽泛采纳计算机控制。计算机对滴定过程中的数据自动收集、办理,并利用滴定反响化学计量点前后电位突变的特征,自动找寻滴定终点、控制滴定速度,抵达终点时自动停止滴定,所以更为自动和快速。图19自动电位滴定装置滴定种类及指示电极的选择酸碱滴定:能够进行某些极弱酸(碱)的滴定。指示剂法滴定弱酸碱时,正确滴定的要求必需≥10-8,而电位法只需大于等于10-10;电位法所用的指示电极为pH玻璃电极。氧化复原滴定:指示剂法正确滴定的要求是滴定反响中,氧化剂和复原剂的标准电位之差必需△φo≥0.36V(n=1),而电位法只需大于等于0.2V,应用范围广;电位法采纳的指示电极一般采纳零类电极(常用Pt电极)。络合滴定:指示剂法正确滴定的要求是,滴定反响生成络合物的稳固常数必需是
≥6,而电位法可用于稳固常数更小的络合物;电位法所用的指示电极一般有两种,一种是
Pt
电极或某种离子选择电极,另一种倒是
Hg电极(其实是第三类电极)。积淀滴定:电位法应用比指示剂法宽泛,特别是某些在指示剂滴定法中难找到指示剂或难以进行选择滴定的混淆物系统,电位法常常能够进行;电位法所用的指示电极主假如离子选择电极,也可用银电极或汞电极。练习题与思虑题电位剖析法的理论基础是什么?它能够分红哪两类剖析方法?它们各有何特色?以氟离子选择性电极为例,画出离子选择电极的基本结构图,并指出各部分的名称。何谓扩散电位和道南电位(相间电位)?写出离子选择电极膜电位和电极电位的能斯特方程式。试述pH玻璃电极的响应机理。解说pH的操作性适用定义。5.何谓ISE的不对称电位?在使用pH玻璃电极时,如何减少不对称电位对pH丈量的影响?气敏电极在结构上与一般的ISE有何不同?其原理如何?何谓ISE的电位选择系数?它在电位剖析中有何重要意义?写出有扰乱离子存在下的能斯特方程的扩大式。何谓总离子强度调理缓冲剂?它的作用是什么?电位滴定的终点确立有哪几种方法?10.计算时以下电池的电动势,并注明电极的正负:已知:答案:0.081V11.硫化银膜电极以银丝为内参比电极,0.0100mol/L硝酸银为内参比溶液,计算该电极在碱性溶液中的电极电位已知:答案:-0.522V12.测定以下电池:pH玻璃电极|pH=5.00的溶液|SCE,获取电动势为0.2018V;而测定另一未知酸度的溶液时,电动势0.2366V。电极的实质响应斜率为58.0mV/pH。计算未知液得pH。答案:5.6冠醚中性载体膜钾电极和饱和甘汞电极(以醋酸锂为盐桥)构成丈量电池为:K+-ISE|丈量溶液|SCE当丈量溶液分别为溶液和溶液时,测得电动势为-88.8mV和58.2mV,若电极的响应斜率为58.0mV/pK时,计算。答案:14.用固定扰乱法测选择电极对的电位选择系数。在浓度为的一系列不同浓度的溶液中,测得以下数据。请用作图法求30.02.0-26.0-45.0-47.0答案:0.2115.选择电极的,当它用于测定pH为6.0且含有溶液中的的时,预计方法的相对偏差有多大?答案:20%16.用选择电极测定水样中的。取25.00mL水样,加入25.00mLTISAB溶液,测得电位值为0.1372V(VsSCE);再加入的标准溶液1.00mL,测得电位值为0.1170V,电极的响应斜率为58.0mV/pF。计算水样中的浓度(需考虑稀释效应)答案:17.为了测定Cu(Ⅱ)-EDTA络和物的稳固常数,组装了以下电池:测得该电池的电动势为0.277V,请计算络和物的。答案:18.正确移取测得其电位值为
50.00mL含的试液,经碱化后(若体积不变)用气敏氨电极测得其电位为-80.1mV。若加的标准溶液0.50mL,测得电位值为-96.1mV。而后在此溶液中再加入离子强度调理剂50.00mL。-78.3mV。计算试液中的浓度为多少(以表示)。答案:19.用氰离子选择电极测定和混淆液中。该电极合用的测得电位值为-251.8mV。而后用固体试剂调理试液至pH=4(此时,mL。若向pH=4的该试液中再加的斜率为,。请计算混淆试液中
pH范围为11-12。现移取试液100.0mL,在pH为12时完好以HCN形式存在);测得电位值为-235.0标准溶液,测得电位值为-291.0mV。已知该电极的响应的浓度。答案:在以下构成的电池形式中:用
的
,
溶液滴定
,
的
KI
溶液。已知,碘电极的响应斜率为。请计算滴定开始和终点时的电动势。
,答案:0.258V、-0.085V采纳以下反响进行电位滴准时,应采纳什么指示电极?并写出滴定方程式。电解与库仑剖析法要点与难点1.分解电压、析出电位、极化和超电压的观点及有关关系2.控制电位电解时离子析出次序及电解分别3.法拉第定律的意义及有关应用和运算4.电流效率的意义及要求电解与库仑剖析电解剖析法是一种经典的电化学剖析法,它包含两种内容:电重量剖析法――经过电解后直接称量电极上被测物质的质量进行剖析的,常用于高含量物质的剖析;电解剖析法――控制必定的电解条件进行电解以达到不同物质的分别。库仑剖析法也是成立在电解过程上的剖析法,它是经过丈量电解过程所耗费的电量来进行剖析的,主要用于微量或痕量物质的分析。电解装置和电解现象电解装置主要由电解池(包含电极、电解溶液及搅拌器)、外加电压装置(分压器)
及显示仪器三部分,如图
1所示。图1电解装置电解是利用外面电能使化学反响向非自觉方向进行的过程。在电解池的两电极上施加的直流电压达到必定值时,电极上就发生氧化复原反响,电解池中(及回路)就有电流经过,这个过程称为电解。以在0.1mol/LHNO3介质中电解0.1mol/LCuSO4为例,当挪动分压器的滑线电阻,使施加到两铂电极上的电压(V)达2+到必定值时,电解就发生,即电极反响发生:与外电源负极连结的Pt电极(此时也是负极)上Cu被复原,此电极为阴极:Cu2+
+2e
→
Cu与外电源正极连结达的Pt电极(此时也是正极)上有气体O2产生,此电极为阳极:2H2O此时在外线路的电表上能够看到有电流(i)经过,若加大外电压,则电流快速上
→4H++O2↑+4e升。i-V关系曲线为图2所示图2电解过程电流-电压曲线应当注意到:电解所产生的电流(电解电流)是与电极上的反响亲密有关的,电流出入电解池是经过电极反响来达成的,与电流经过一般的导体有实质的不同。这是电解的一大特色。分解电压和析出电位图2的i-V曲线中,AB段为剩余电流,此时还没有察看到电极反响的明显发生,主假如充电电流,当抵达必定的外加电(B点)时,电极反响开始发生,产生了电所对应的电压,叫做分解电压V分。V
压V解电流,并跟着V的增大而快速上涨为BC直线,BC线的延长线与i=0的V轴交点分定义为:被电解物质能在电极上快速、连续不停地进行电极反响所需的最小外加电压。
D为何电解的发生需要分解电压呢?电解的另一大特色是,电解一开始,就为其建立了对峙面――反电解,即电解一开始产生了一个与外加电压极性相反的反电压,阻挡电解的进行,只有不停的战胜反电压,电解才可进行和连续。――观察电解CuSO4溶液的进度,两支相同的一个很小电压时,在最先的瞬时,就会有极少许的
Pt
电极插入溶液,当外加电压为零时,电极不发生任何变化;当两电极外加Cu和O2分别在阴极和阳极上产生并附着,因此使本来完好相同的电极,变为
Cu电极和氧电极,构成一个原电池,产生一个与外电压极性相反的电动势,在电解池中,此电动势称为反
电动势,它阻挡电解的连续进行,假如除掉外加电压,两电极短路,就产生反电解,
Cu从头被氧化成
Cu2+,O2从头被复原成
H2O。理论上,只有外加电压增添到能战胜反电动势时,电解方可进行,此时的外加电压叫做理论分解电压
V分(理),明显:("平"指均衡电位)分解电压是对电池整体而言的,若对某工作电极的电极反响来说,还可用析出电位来表达。假如电解池中再配上一支参比电极,在不同外加电压下监测工作电极的电流,并丈量电解电流,绘制i-φ曲线,相同能够得出,只有工作电极的电位达到某一值时,电极反响才发生,这个电位称为析出电位,φ析。φ析定义为:能使物质在阴极快速、连续不停的进行电极反响而复原所需的最正的阴极电位,或在阳极被氧化所需的最负的阳极电位。明显,若外加电压使阴极电位比阴极析出电位更负一点,或阳极电位比阳极析出电位改正一点,电极反响就能快速、连续不停地进行,理论上,析出电位等于电极的均衡电位,称为理论析出电位φ析(理),即:所以,超电压和电解方程式在导论一章中已指出,电流经过电解池时,因为两电极会发生极化并由此产生过电位,总过电位
η总可表示为:也称为总过电位和超电压
所以,实质的分解电压
V分,除了要战胜电解池的反电动势外,还应战胜超电压,所以行后,电解池路上有电流
:
i经过,外电源所施加的电压
V外还一部分用于回路电阻
R的电压降,所以,总外加电压
当电解进V外为:―――――电解方程式影响电极过电位的要素:++η要大。电流密度――电流密度愈大,η也愈大。温度――温度高升,会使离子的扩散速度和电极反响速度加速,故η降低。电极反响析出物的状态――析出气体的η大,因为气领会在电极表面聚成气泡附在电极表面,减少电极与溶液的接触面积,阻挡扩散及反响。析出物能与电极形成金属齐(如汞齐)的η较小。控制电流电解剖析法(也叫恒电流电解剖析法)图3恒电流电解装置控制电流电解剖析法装置如图3所示(阴极电解)。该法的最大特色是,以大面积的铂网为阴极,以螺旋状的铂丝作为阳极,并连结到马达上作搅拌器,装置还常常配有加热设施。电解剖析一开始就施加较高的外加电压,产生较为稳固的电解电流(一般3A以下)。跟着电解的进行,电流会衰减,因此需不停增添外加电压,以保持电流的基本稳固。经一段时间的电解,待待测物质完好析出在铂网上后,拿出铂网,洗净,烘干并称重。这个方法的仪器简单,剖析速度快,正确度高。剖析的正确度在很大程度上取决于电分析出物的性状。析出物必需纯净并坚固附着在电极上,以防备洗、烘、称时零落。采纳大面积的铂网能够降低电流密度,充分搅拌能够使析出物均匀,采纳络合性的电解液能够使电极反响平和,析出物较致密。这个方法的最大弊端是选择性较差,因为外加电压较大,常常一种金属离子未完好析出时,另一种金属离子就在电极上析出,共存离子扰乱较为突出,并且外加电压加大到必定程度时,就惹起放H2反响。所以,这个方法合用于溶液中仅含一种比H+更易复原的金属离子的测定(即电动序表中氢后的金属)或作电动势在氢从前和以后金属的分别。改变介质条件,如在碱性或络合剂存在下的介质中电解,能够扩大应用范围。经过加入去极剂(或称电位缓冲剂),可使外加电压加大到某一程度时,工作电极的电位保持不变,防备扰乱的电极反应的发生。如在2+2+混淆液中,为防备2+在分别Cu时析出,可加入-作为阴极去极剂,-在阴极上复原:Cu,PbPbNO3NO3复原电位比2+-复原在2+从前,且-量又大,所以Pb不会析出。Pb正,NO3PbNO3控制电位电解剖析法装置及电解过程控制电位电解剖析是在控制工作电极的电位为必定值的条件下进行电解的方法,在装置上比起图3.3恒电流电解剖析法多了一个控制和丈量工作电极电位的设施。所以,采纳三电极系统,由工作电极和对电极与外加电压电源构成电解回路,而工作电极和参比电极连结电子伏特计构成工作电极电位的鉴测回路。4电流与时间关系剖析时,依据被电解物质完好析出时所应控制的电位,选择适合的外加电压加到电极上。因为电解刚开始时,离子浓度很大,所以电解电流也很大,电解速度很快,跟着电解的进行,离子浓度降低很快,电流急剧降落,当电流趋近于零时,表示电解基本完全。电解电流i随电解时间t变化的曲线如图4所示。如电解时,仅有一种物质以100%的电流效率电解,则i、t的关系为:式中:it为电解时间为t(min)的电流,io为开端电流,k(min-1)为与电极及溶液性质有关的常数:式中:D为扩散系数(cm2/s),A为电极表面积(cm2),V为溶液体积(cm3),δ为扩散层厚度(cm)可见,若要缩短剖析时间,应增大K值,就要求电极表面积要大,溶液体积要小,提升溶液的温度及优秀的搅拌以增大扩散系数和降低扩散层厚度。控制电位电解分别时,离子析出的序次及完好程度电解测定某一离子时,一定考虑其余共存离子的共堆积问题,而利用控制电位进行混淆离子的分别和剖析,也一定考虑离子析出的序次及分别完好度的问题。不同离子析出电位的差异决定了它们电分析出的序次。在阴极上,φ阴析愈正者,愈易复原,则先析出;在阳极上,φ阳析愈负者,愈易氧化,则先析出(或溶解)。两离子析出电位的差异△φ析决定了其可否经过控制电位电解达到完好分别。如电解Cu2+和Ag+混淆溶液中,因为△φ析差别大,故可分别;而Pb2+,Sn2+的△φ析小,故难以进行电解分别。在电解剖析中,往常把离子的浓度降至初始浓度的10-5~10-6倍时,视为电分析出完好。所以关于两混淆离子要能经过控制电位电解达到完好分别,其析出电位之差(V)(即)。所以,当阴极电压控制在0.337-0.385V之间,电解结束时,Ag+能够完好复原析出而Cu2+仍留在溶液中,达到完好分别(若要求使降至10-8mol·L-1状况如何?)假如要连续电解Cu2+,则改换阴极,降低阴极的控制电位连续电解。要使Cu2+完好复原析出(降至10-6mol·L-1),应控制的阴极电位为:5汞电极电解装置库仑剖析法的基根源理1.法拉第(Faraday)定律数学表示式为:式中:m为电极上析出物质的质量,M为物质的摩尔质量,n为电极反响的电子转移数,Q为通过电解池的电量,F为Faraday常数(96487库仑/摩尔)其式有两层含义:――关于电解同一物质,m∝Q――关于电解不同物质,当Q相同时,m∝M/n2.电流效率关于库仑剖析来说,经过电解池的电量应当所有用于丈量物质的电极反响,即待测物质的电流效率应100%,这是库仑剖析的先决条件。即电极反响是单调的,没有其余副反响发生。影响电流效率的要素:溶剂的电解――电解一般在水溶液中进行,所以溶剂的电解就是水的电解,即阴极放氢、阳极放氧的反响。防备水电解的方法是控制适合的电解电位、控制适合的pH值及选择过电位高的电极。杂质的电解――电解溶液中的杂质可能是试剂的引入或样品中的共存物质,在控制的电位下会电解而扰乱,除去的方法是试剂提纯或空白扣除,试液中杂质的分别或遮蔽。溶液中溶解氧的电解――溶解氧可在电极上复原,。除去的方法可向电解溶液中通高纯N2驱氧或在中性、弱碱性溶液中加入Na2SO3除氧。电极参加电极反响――有的惰性电极,如Pt电极,氧化电位很高,不易被氧化,但若有络合剂存在(如大量Cl-),使其电位降低而可能被氧化。防备方法是改变电解溶液的构成或改换电极。电解产物的再反响――可能是一个电极上的产物与另一个电极上的产物反响或电极反响产物与溶液中某物质再反响。如阴极复原Cr3+为Cr2+时,Cr2+会被H+氧化又从头生成Cr3+。战胜的方法是改变电解溶液。以上的影响要素中溶剂和杂质的电解是主要的。库仑剖析的方法控制电位库仑剖析法●方法原理及装置:成立在控制电位电解过程的库仑剖析法称为控制电位库仑剖析法。即在控制必定电位下,使被测物质以100%的电流效率进行电解,当电解电流趋于零时,表示该物质已被电解完好,经过丈量所耗费的电量而获取被测物质的量。控制电位库仑剖析的装置比起控制电位电解剖析多了一个电量丈量部分,如图6所示。图6控制电位库仑法的基本装置图7银库仑计●电量的丈量:★重量库仑计――主要有银库仑计。结构如图7所示。以铂坩埚为阴极,银棒为阳极,用多孔瓷管把两极分开,坩埚内盛有1-2mol·L-1的AgNO3溶液。串连到电解回路上,电解时发生以下反响:阳极阴极电解结束后,称量坩埚的增重,由析出银的量算出所耗费的电量。别的还有钼库仑计、铜库仑计、汞库仑计等。★气体库仑计――有氢氧和氮氧气体库仑计图8氢氧库仑计氢氧库仑计如图8所示。装有-124Pt片电极,串通到电解回路中,电0.5mol·LKSO溶液的电解管置于恒温水浴中,管下方焊上两解时,两Pt电极上分别析出H2和O2阳极阴极电解结束后,刻度管电解前后液面之差为电分析出H2和O2混淆气体的体积。从电极反响式及气体定律可知,在标准状况(273K,760mmHg压力)下,经过每法拉第电量(96487库仑)可产生16800mL混淆气体。(或每库仑可产生0.1741mL气体)。所以可算出电解所耗费的总电量。★化学库仑计――也称滴定库仑计其结构如图9所示,杯内盛0.03mol·L-1的KBr和0.2mol·L-1的K2SO4。电解发生时,电极反响为:阳极阴极电解结束时,用标准酸溶液滴定电解生成的-OH的量,因此可算出耗费的总电量。图9化学库仑计★电子积分仪――库仑剖析中的电量为采纳电子线路积分总电量并直接从仪表中读出,甚为方便、正确。别的,可用作图法求电量――控制电位库仑剖析中的电流随时间而衰减的函数式为:,则当t较大时,kt>3,10-kt能够忽视所以而电解中测定n对t、it数值,作~t曲线,其斜率为-K,截距为,代入前式即可求得Q。●控制电位库仑剖析法的特色和应用:★该法是丈量电量而非称量,所以可用于溶液中均相电极反响或电极反响析出物不易称量的测定,对有机物测定和生化剖析及研究上有较独到的应用。★剖析的敏捷度、正确度都较高,用于微量甚至痕量剖析,可测定级的物质,偏差可达0.1~0.5%。★可用于电极过程及反响机理的研究,如测定反响的电子转移数、扩散系数等。★仪器结构相对较为复杂,杂质及背景电流影响不易除去,电解时间较长。2.控制电流库仑剖析法--库仑滴定法●方法原理及装置库仑滴定法是用恒定的电流经过电解池,以100%的电流效率电解产生一种物质(称为"电生滴定剂")与被测物质进行定量反响,当反响抵达化学计量点时,由耗费的电量(it)算得被测物质的量。可见,它与一般滴定剖析方法的不同在于:滴定剂是由电生的,而不是由滴定管加入,其计量标准量为时间及电流(或Q),而不是一般滴定法的标准溶液的浓度及体积。库仑滴定法的装置除了电解池外,还需有恒电流源,计时器及终点指示装置。图10为其表示图。图10库伦滴定装置图11永停终点法装置●指示终点的方法★指示剂法――是简易、经济适用的方法。指示剂一定是在电解条件下的非电活性物质。指示剂的变色范围一般较宽,指示终点不够敏锐,故偏差较大。★电位法――与电位滴定法指示终点的原理相同,采纳适合的指示电极来指示滴定终点前后电位的突变,其滴定曲线可用电位(或pH)对电解时间的关系表示。★双指示电极(双Pt电极)电流指示法――也称永停(或死停)终点法,其装置如图11所示,在两支大小相同的Pt电极上加上一个50~200mV的小电压,并串通上敏捷检流计,这样只有在电解池中可逆电对的氧化态和原还态同时存在时,指示系统回路上才有电流经过,而电流的大小取决于氧化态和复原态浓度的比值。当滴定抵达终点时,因为电解液中或许本来的可逆电抵消逝,或许新产生可逆电对,使指示回路的电流停止变化或快速变化。如在Ce3+和Fe2+溶液中,电生Ce4+滴定和Fe2+,指示回路中电流随时间的变化曲线如图12所示;在KBr和AsO33-溶液中,电Br2滴定AsO33-,i~t曲线如图13所示。库仑剖析的方法●库仑滴定法的特色和应用应用较宽泛,凡能与电生滴定剂起定量反响的物质均可测定,常有的剖析见表1和表2表1应用酸碱、积淀及络合反响的库仑滴定法被测物产生滴定剂的电极反响滴定反响质酸2H2O+2e-=H2+2OH--=H2OOH+H碱2H2O=O2+4H++4e-H++OH-=H2O卤离子Ag=Ag++e-Ag++X-=AgX↓硫醇Ag=Ag++e-Ag++RSH=AgSH↓+H+氯离子2Hg=Hg2++2e-2++2Cl-=Hg2Cl2↓2Hg22+Fe(CN)3-+e=Fe(CN)4-4-+3Zn2++2K+=K2Zn3[Fe(CN)6]2↓Zn662Fe(CN)6Ca2+,2+2-++2e-=Hg+2NH3+HY3-(Y4-为EDTACuHgNHY+NH43HY3-+Ca2+=CaY2-+H+Zn2+,离子)Pb2+表2库仑滴定法产
温馨提示
- 1. 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
- 2. 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
- 3. 本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
- 4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
- 5. 人人文库网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
- 6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
- 7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
最新文档
- 硅藻泥室内墙面施工方案
- 装企erp 实施方案
- 团县委禁毒推进工作方案
- 村里的电话核查工作方案
- 全员出动后续工作方案
- 外卖公关运营方案模板
- 物流业智慧仓储管理方案
- 智能制造数字化转型实施方案
- 2026年建筑工程施工分包合同三篇
- 【绿色消费行为的外部情景因素的研究文献综述5000字】
- 虹口区2024-2025学年下学期期末考试六年级数学试卷及答案(上海新教材沪教版)
- 2025年中国邮政集团有限公司上海市分公司人员招聘笔试备考试题及参考答案详解1套
- 2025年湖南省高考物理试卷真题(含答案解析)
- 高速监控管理制度
- 水利工程可行性研究报告审查要点
- T-ZSA 288-2024 餐饮设备智能烹饪机器人系统通.用技术要求
- 反诈辅警年度考核个人总结
- 我的家乡定西
- IE-7大手法之人机分析
- 2024年高考湖南卷物理真题(解析版)
- 电影叙事与美学智慧树知到期末考试答案章节答案2024年南开大学
评论
0/150
提交评论